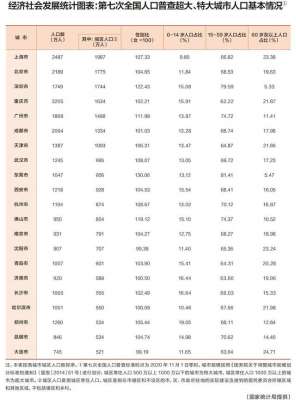
阴离子(一文掌握富锂正极中阴离子氧化还原行为)
来源:峰值财经 发布时间:2023-05-04 浏览量:次

【研究背景】
在锂离子电池正极材料中,富锂正极材料以其较高的比容量获得了极高的关注和广泛的研究。富锂正极材料中独特的超晶格结构和阴离子氧化还原反应也为氧化物层状正极的基础研究提供了很好的载体。然而,具体的反应机理尚不明确,人们对于其容量和电压的衰减机制认识尚存在不完整甚至是矛盾的,这阻碍了富锂正极材料的应用,另一方面也不利于对正极材料作用机制的深入挖掘和基础研究。
基于此,首尔大学Yong-Mook Kang团队在Advanced Materials ,上发表题为“A Mechanistic Insight into the Oxygen Redox of Li-Rich Layered Cathodes and their Related Electronic/Atomic Behaviors Upon Cycling”的综述论文,详细总结了目前富锂正极的研究进展及晶格氧阴离子氧化还原反应的作用机制,以及目前针对其性能衰减的改性机制。本文对丰富阴离子氧化还原作用机理及富锂正极材料改性设计提供了较为全面的总结和参考。
【图文导读】
原子结构
在LiMO2 (M:金属离子)中,Li和过渡金属交替占据着不同的八面体位置,这导致了其具有六方晶系的O3型结构(图1a)。在相同的氧骨架结构中,过剩的Li将部分替代TMs,使其转变为单斜结构(图1b)。过量的Li渗透到TM层中,单斜结构特征峰介于20°和30°之间(图1d),这是Li+和Mn4+之间的电荷和尺寸的差异(图1c)导致了LiM6蜂窝状排列结构的形成(图1d)。通过引入Li/Mn无序,计算得到的XRD图谱保持了主结构的Bragg峰, (图1e)。图1f显示了随着叠加无序度的增加,低角度衍射峰消失。

图1富锂正极中两相的晶体结构及XRD衍射谱。
电子结构和阴离子氧化还原
对于具有化学计量比Li/TM的层状金属氧化物,氧化物离子由3个TM离子和3个Li离子配位,如图2a所示。由于费米能级位于具有M的轨道中,LiMO2的容量源于M氧化还原,即阳离子氧化还原(图2c)。在富含Li的材料中,氧化物离子由两个TM离子和四个Li-O-Li构型的Li离子配位(图2b),这是由于Li占据了TM位点。由于Li-2s轨道和O-2p轨道之间的能量差较大,杂化受限,形成了非键(NB)的O 2p轨道,其能级位于具有M的轨道和费米能级附近的O特征轨道之间,该能级允许从O 2p NB态中去除电子(图2d),即阴离子氧化还原。受当前阴离子氧化还原理论的启发,人们采取了一种更直接的方法,随着Li/M和O/M比的增加,Li-O-Li构型数量增加,从而在费米能级附近形成更多的O 2p NB态(图2e)。

图2 改不同结构中O的配位环境、轨道杂化状态、及对应的能带结构。
阴离子氧化还原研究进展
Okubo等人针对氧化物的八面体构型,推断OM2Li4配位具有C2v对称性,允许π型重叠,形成b1/b1*轨道(图2b)。电子去除后,较大的b1/b1*分裂稳定了晶格氧的氧化(图3a)。根据计算结果,当E(M t2g)>E(O 2p)时,b1*具有M的特征,而O特征在相反的情况下更强(图3b)。利用具有C2对称性的高共价型Li2RhO3材料,可计算推导出σ、π和O 2p NB态的形成(图3c)。Sudayama等人揭示了O氧化还原的稳定机制(图3e)。π中心的离域降低了氧化势和带电态的自由能(图3f)。这些特性提高了晶格氧氧化的可逆性和结构稳定性,从而降低了材料的电压滞后。

图3 b1/b1*的轨道杂化能级图、能量分布、不同SOC下电子转移情况及对应的Mn溶出、O损失过程。
Lai等人比较了Mg/Zn取代LixMnO2 (LMMO)和NaxMnO2 (NMMO)的阴离子氧化还原行为。计算的PDOS显示,LMMO和NMMO都有接近费米能级的O 2p轨道(图4a)。然而,LMMO的容量较低,这表明其没有氧氧化还原(图4b)。对比两种氧,一种靠近阳离子空位(OA),一种远离空位(OB),OA的PDOS在费米能级以上显示出更多的OA电子态,说明OA具有氧化还原性质,而OB只有轻微的差异(图4c)。Lee等人利用NMC材料结合mRIXS表征获得了晶格氧被氧化的实验证据(图4d中),然而目前的理论基础很难找到合适的解释。结合O-K边和Mn-L的mRIXS-iPFY的分析表明,在第一次充电期间没有可逆氧氧化还原(图4d右),而随后的循环仅与Mn3+/4+氧化还原相关,这一点类似于传统正极材料。

图4 NMMO和LMMO的O 2p和Mn 3d轨道的PDOS分布、充放电曲线及同步辐射成像图。
阴离子氧化还原反应的可逆性
在充电初期,电子转移仅发生在填充的LHB中,对应于单波段阳离子氧化还原(图5a左)。高价的TM(如V, Mo, Cr)会重组形成稳定的TM=O多重键(<2 Å)(图5b下)。中间态和末态的TM Mott-Hubbard体系涉及大部分4d和5d过渡金属氧化物,其中LHB与O 2p NB态重叠,这导致了阳离子氧化还原,然后是阴离子氧化还原,伴随着Jahn-Teller畸变或Peierls扭曲的氧骨架重组(图5a中)。电荷转移返回到一个单波段氧化还原过程,并从O 2p NB波段脱出电子(图5a右)。这种情况包括富Li的3d TM氧化物,其中O 2p NB态和M d态之间没有重叠,在局部的O 2p NB态中产生高度不稳定的空穴。它们的重排形成O-O种(图5b顶部)和O 2p NB带分裂为σ*、π*、π和σ带(图5c)。在Li2MO3的中间态下,氧化还原活性M将补偿锂离子在脱除时的电荷转移,从而将每个氧的电子空穴减少到1/3,提升其稳定性(图5d-a)。在Li2M’O3型电荷转移电极中,由于M’是氧化还原非活性的,每个氧在完全脱除后会形成2/3个空穴,这就导致电压滞后和容量衰减(图5d-d)。如果O2释放激活了最初在第一个循环中不活跃的额外TM,则将出现电压滞后和容量损失(图5d-c)。

图5 Mott-Hubbard,中间和电荷转移系统的能带结构、O氧化后结构局部重组和对应的能带结构及O 2p NB态的重组及对应的充放电曲线。
电压滞后现象
为了区分阳离子还原氧化和阴离子氧化还原对电压滞后的贡献,研究人员进行了恒流充放电循环,逐渐降低或增加上充电和下放电截止电压(图6a) 。实验结果表明,在较窄的电压窗口内,电压滞后现象减小,这证明了氧化还原的强烈影响。Tsuchimoto等人研究了Na2Mn3O7低电压滞后的原因(图6b),他们通过测量磁化率证明了可逆的O2- /O-氧化还原电对的存在。阳离子/阴离子氧化还原的反转也丰富了电压滞后的潜在机制。描述了氧化还原过程按照TM氧化→O氧化→TM还原→O还原的顺序进行,其中O还原发生在低电位(图6c)。当σ*轨道能级高于Mn d能级时,TM还原和O还原发生反转,使氧的氧化还原电位发生剧烈变化(图6d)。Assat等人提出了一个综合热力学和动力学步骤来解释阴离子氧化还原和电压滞后。他们提出了一个多步骤的反应机制,灵感来自分子机器,用以解释阴离子氧化还原的滞后现象(图6e)。当反应回到放电时,反应的中间产物将首先通过LMCT从氧弛豫到Fe4+,形成Fe3+-O(2-n)-,随后进行氧还原,从而产生较大的电压滞后(图6f)。Ni在高电位平台区发生部分还原,在放电过程中, Ni3+/4+组分首先还原为Ni2+,同时发生O还原(图6g)。Ni和Fe的不同之处在于Ni在放电开始时就可以诱导阳离子还原(图6h)。

图6 富锂NCM正极材料充放电曲线、不同阶段的O反应,能量变化及对应的容量贡献占比。

图7 富锂正极循环过程中的电压衰减及结构重排、相变、TM活性中心的容量占比、三维成像示意图及晶面滑移情况。
电压衰减
电压衰减是在相同的充电状态下,循环过程中材料的电压逐渐下降的现象(图7a)。Gu等证明了尖晶石的形成在Li化学计量区和多重结构转变,如非晶化和层状到尖晶石的转变出现在富Li区域(图7b)。进一步的循环证明了无序岩盐相的大量生长,相变从层状结构→缺陷尖晶石相→无序岩盐发生(图7c)。Hu等人发现,在富含Li的NCM中,O2释放后将激活低电位的Mn和Co氧化还原反应(图7d)。在3D可视化结果中,Xu等证明了早期循环中孤立空穴的形成,在20次循环后发展成一个大的孔隙网络(图7e)。Singer等人利用原位布拉格相干衍射成像(BCDI)观察了Li1.2Ni0.133Mn0.533Co0.133O2纳米颗粒循环时部分位错的成核(图7f),堆叠层错的排布方式变化也会导致电压衰减。

图8 富锂正极充放电曲线及电化学原位质谱、衰减机理示意图、恒电流间歇滴定测试性能图。
首次循环不可逆现象
首次循环作用机制由Luo等人首先提出的。他们发现在高电压平台的O释放贡献了9%的容量,而4.5 V以上的容量几乎全部来自O释放。实验结果与理论计算相结合,证明TM迁移是由于O的氧化引起的局部O构型的变化(图8b),并导致O氧化还原电位的偏移,从而导致第一循环充放电极化较大。Li+在还原后占据了新的TM空位,完全改变了氧化离子的局部环境,导致放电时产生了较大的电压差(图8c)。因此,第一个电压平台可以归因于从均匀形态到纳米结构形态的转变,相关的不可逆阳离子无序描述了放电时平台的消失(图8d)。表面无序尖晶石经过可逆转化为岩盐结构(图8e)。在常规电压窗口(4.8-2.0 V)内,他们观察到氧化程度和不可逆容量之间有很强的相关性(图8f)。P′相的容量恢复对应GITT测试中严重的极化,表明Mn迁移导致的Li+扩散缓慢是不可逆容量产生的原因(图8f)。

图9 富锂正极材料的表面改性机制、内建电场示意图、Li含量梯度分布设计及对应的能带图。
抑制晶格氧的析出
利用NH4F处理材料将表面部分转化为尖晶石样相,形成纳米异质结,促进了材料的活化(图9a)。Zhang等人将反位尖晶石MgTiO4包覆在Li1.2Mn0.54Ni0.13Co0.13O2上,阻断氧气释放(图9b)。结果表明,由于包覆的反位尖晶石表面的介电特性,形成了反向电场(图9c),这抑制了O2的释放。根据第一性原理计算可知,氧空位通过四面体位时迁移势垒降低,这也激活了Li扩散(图9d)。此外,通过气体处理得到预活化的Li2MnO3,阻止了第一次充电时的O2的形成,如图9e所示。由于副反应的控制和电极/电解质界面电阻的降低,O2释放的减少形成了较薄的SEI层。通过熔融MoO3对Li1.20Mn0.48Ni0.16Co0.16O2进行高温浸出(图9f),此举增加了从表面到本体的Li浓度梯度。由于Li过剩的环境,Li-O-Li构型在本体中触发阴离子氧化还原,从而降低了材料表面的Li缺陷(图9g)。

图10 Ru掺杂改性抑制O损失、Li/Ru混排机制、HADDF-STEM图及电化学原位质谱。
抑制过渡金属离子迁移
Mozorov等人使用Ru在Li1.2Ni0.2Mn0.6O2中替换了少量的这两种3d过渡金属。与之前的研究相似,增加Ru抑制了第一次充电时的高电位氧的氧化平台,OEMS在第一次循环中无O2释放(图10a从左到右)。相关的表面致密化在第20个循环时被抑制(图10b),低电位Mn3+/4+对的激活也被抑制,而Mn k边XANES光谱中没有峰移(图10c)。图10d显示了两个具有轴对称氧中心的共享边八面体的有序排列。由于这种对称性,氧化氧沿着O-O轴的方向靠近Ru(图10e左)。然而,当Li/Ru无序分布破坏了轴对称时,O-Ru-O构型便会优先形成(图10e右)。无序的Li2RuO3表现出接近96%的高循环容量保持率和0.07 V的低电压衰减。拉曼光谱显示不存在(O2)拉伸,而dems也显示不存在O2产生(图10f)。

图11 Mg2+掺杂作用机理、过渡金属离子迁移路径及缺陷产生机制。
Liu等在富Li NCM层间Li位点掺杂Mg2+,结果发现材料没有发生明显的结构变化。结果显示,电化学非活性的Mg2+在循环时依然保留在Li位点。因此,TM迁移路径被Mg2+和TM的静电斥力所阻断,从而抑制了结构转变(图11a)。八面体位点的TMs作为刚性支柱,而四面体位点的TMs作为自适应支柱,在锂化后移动回TM层(图11b)。与传统的O3型结构相比,O2型结构的静电斥力更大,可将TM保持在四面体位置 (图11c)。掺杂Na离子也能改变层间环境。Na离子一般比Li离子大1.16 Å半径(~ 0.74 Å)。因此,Na在层间的插入扩大了层间间距,从而抑制了TM的迁移(图11d)。House等人实验证明,第一次充放电循环后仍保持层状有序结构,而蜂窝有序超晶格结构则发生面内无序重组(图10e)
【总结和展望】
本文总结了阴离子氧化还原的不同作用机制,并综述了富锂层状正极的实际问题和解决方案。尽管学界在富锂材料和阴离子氧化还原方面取得了可观的研究成果,但仍有许多问题尚未解决。不仅如此,富锂正极的电池组件适配性也需要严格考虑。需要仔细考虑通过控制电压窗口来限制材料可逆脱嵌锂的量。此外,目前的电池制造程序包括活化过程,该过程会激发产气,可以采用恒流恒压(CCCV)方法去除O2,从而以牺牲氧气氧化还原的可逆能力为代价,获得更好的循环性能。从安全角度来看,电池单体/电池组的合适设计则是另一种选择。以上都有利于富锂正极材料的高效利用。
Seongkoo Kang, Dayeon Choi, Hakwoo Lee, Byungjin Choi, Yong-Mook Kang. A Mechanistic Insight into the Oxygen Redox of Li-Rich Layered Cathodes and their Related Electronic/Atomic Behaviors Upon Cycling. Adv. Mater., 2023. DOI: 10.1002/adma.202211965
https://onlinelibrary.wiley.com/doi/10.1002/adma.202211965